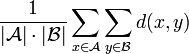UPGMA (Unweighted Pair Group Method with Arithmetic Mean) is a simple agglomerative or hierarchical clustering method used in bioinformatics for the creation of phenetic trees (phenograms). UPGMA assumes a constant rate of evolution (molecular clock hypothesis), and is not a well-regarded method for inferring relationships unless this assumption has been tested and justified for the data set being used. UPGMA was initially designed for use in protein electrophoresis studies, but is currently most often used to produce guide trees for more sophisticated phylogenetic reconstruction algorithms.
The algorithm examines the structure present in a pairwise distance matrix (or a similarity matrix) to then construct a rooted tree (dendrogram).
At each step, the nearest two clusters are combined into a higher-level cluster. The distance between any two clusters A and B is taken to be the average of all distances between pairs of objects "x" in A and "y" in B, that is, the mean distance between elements of each cluster:
The method is generally attributed to Sokal and Michener. Fionn Murtagh found a time optimal O(n2) time algorithm to construct the UPGMA tree.
2. Key Points:
- Sequential clustering algorithm
- Start with things most similar
- Build a composite OTU(Operational Taxonomic Units)
- Distances to this OTU are computed asarithmetic means–
- From new group of OTUs, pick pair withhighest similarity etc.
- Average-linkage clustering
3. UPGMA Visually:
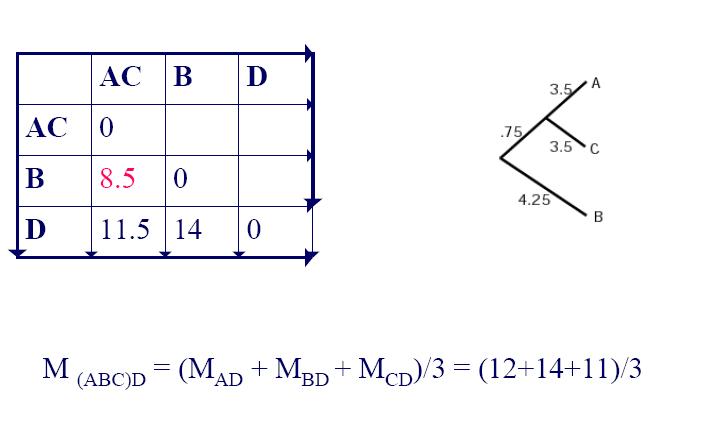
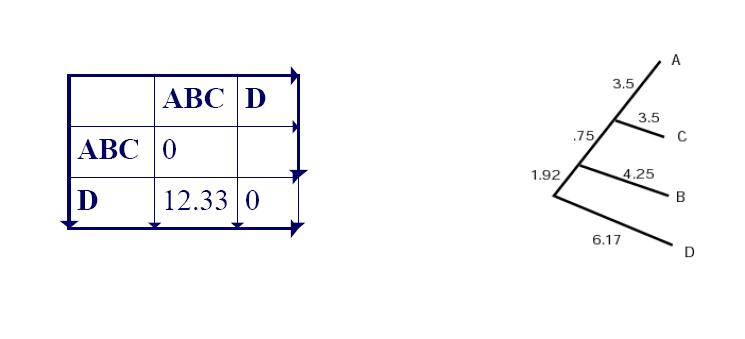
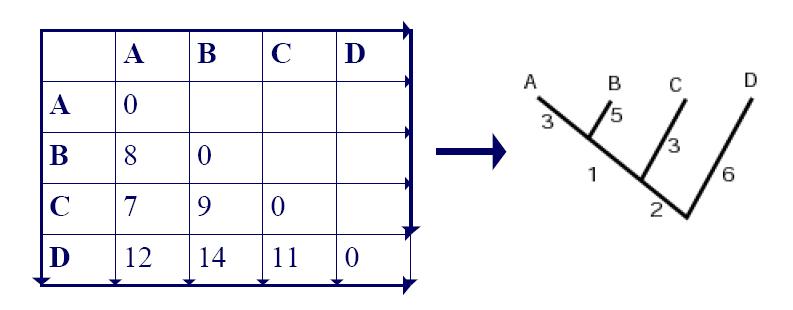
4. UPGMA Example:
5. UPGMA Weaknesses:
In fact, exact fitting tree exists !
- UPGMA assumes that the rates of evolutionare the same among different lineages
- In general, should not use this method forphylogenetic tree reconstruction (unlessbelieve assumption)
- Produces a rooted tree
- As a general clustering method (as wediscussed in an earlier lecture), it is better…