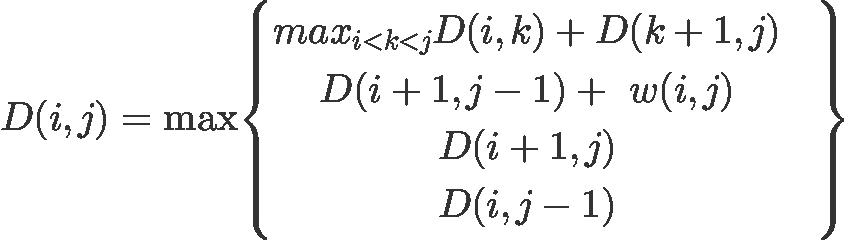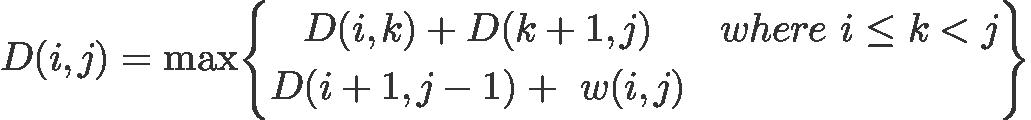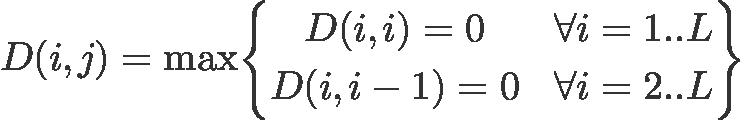Nussinov algorithm is an algorithm to predict possible RNA secondary structure (folding), discovering parts that have complementary sequences. Unlike search algorithms, this method works with the single sequence. It computes the sequence against itself using dynamic programming table, however the matching scores are set so that the letters are treated as "matching" if the corresponding nucleotides would pair. Hence A "matches" U and G "matches" C, but none of the four (AGUC) nucleotides matches itself as two identical nucleotides do not pair.
2. Algorithm:
The algorithm is formally written as
Here w(i,j) = 1 if chars at the positions i and j are complementary (AU, UA, GC, CG). Otherwise this function is 0.
Another formula set that may be simpler is given in and referred as Nussinov-Jacobson algorithm:
With initialization
Nussinov algorithm does not necessary generates the most stable structure and may have scattered matches that are not biologically reasonable. It is too simple to be accurate but has been a stepping stone for more complex algorithms.
3. Key points:
Given an RNA sequence of length L.
Find the structure with the most base pairs
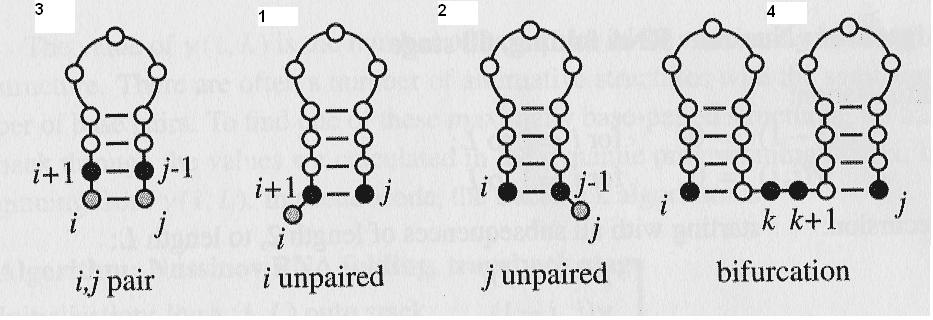
There are four ways of getting the best structure between position i and j from the best structures of the smaller subsequences:
Add unpaired position i onto best structure for subsequence i+1, j
Add unpaired position j onto best structure for subsequence i, j-1
Add i,j pair onto best structure found for subsequence i+1, j-1
Combine two optimal structures i,k and k+1, j
Four Cases
The numbers above each case corresponds to the enumeration on the previous slide.
Compares a sequence against itself in a dynamic programming matrix.
Since structure folds upon itself, it is only necessary to calculate half the matrix.
The value of M[i,j] is the number of base pairs in the maximally base-paired structure.
Four rules for scoring the structure at a particular point.
4. Pseudocode:
Initialization: fill the main diagonal and the diagonal just below it with zeros
Formally, the scoring matrix, M, is initialized:
M[i,i] = 0 for i = 1 to L (main diagonal)
M[i,i-1] = 0 for i = 2 to L (diagonal below main diagonal)
Matrix Fill: Starting with all subsequences of length 2, to length L
do
M[i,j] = max of the following :
M[i+1,j] (base i is hanging off by itself)
M[i,j-1] (base j is hanging off by itself)
M[i+1,j-1] + S(xi, xj) (bases i and j are paired; if xi = complement of xj, then S(xi, xj) = 1; otherwise it is 0)
M[i,j] = MAXi<k<j (M[i,k] + M[k+1,j]) (merging two substructures)
4. Traceback:
5. Pseudoknots:
The algorithm above detects complementary sequences but tells nothing about how the RNA would fold. The usually expected structure ("knots") implies that one independent matching pair of sequences either completely precedes the other or is fully nested within. To emphasize this fact, RNA folding is described using the dot-bracket notation, marking every pairing nucleotide with ( and its matching counterpart with ). Parenthesis in the resulted expression must be balanced, for instance
....(((...)))......(((((......)))))...... - two independent
....(((........(((.....)))...)))......... - one inside another
....(((........(((.....)))..((((...))))....))) - two inside one
Using this rule, it is possible to write a wrapping algorithm that in many cases correctly predicts RNA folding. If these rules are violated, it is said that RNA forms "pseudoknots". Pseudoknots do exist in nature, and more complex algorithms are required to discover them.
6. Alternative Sequences:
A "real world" size RNA may have multiple alternative ways of folding, when either one or another subset of the detected matching sequences are involved. As a rule, RNA folds in the way that is the most "thermodinamicaly stable" - this usually means that the folding case that has the biggest total number of paired nucleotides takes over others.